自然界每一种植物、动物和人类细胞内部,都包含有数以亿计的分子机器。这些分子机器由蛋白质、DNA、RNA及其他配体分子组成。正是这些由生物大分子组成的小型机器,维持着生命的运转和延续。从本质上来讲,生命就是建立在分子层面的结构支撑,以及分子之间的相互作用。因此,准确计算和预测我们体内的生物大分子的原子结构以及分子与分子之间的相互作用,是理解人体疾病机理、理性设计药物、改善人体健康的关键起点。
2024年5月9日,DeepMind与Isomorphic Labs的研究人员在国际顶级杂志《Nature》发表了题为“使用 AlphaFold 3 准确预测生物分子相互作用的结构” (Accurate structure prediction of biomolecular interactions with AlphaFold 3) 的研究论文(图1)。该研究开发了一款新的被称做AlphaFold 3的人工智能(AI)软件,以前所未有的精确度,成功预测了几乎所有生命大分子(蛋白质、DNA、RNA、配体等)的结构和相互作用。

图1. DeepMind和Isomorphic Labs团队在最新一期《Nature》发表AlphaFold3 论文截图。
这种用计算机精准预测和解析蛋白质与其他分子复杂相互作用的能力,有助于为疾病通路、基因组学、治疗靶点、蛋白质工程及合成生物学等领域带来新见解。更重要的是,AlphaFold3 为药物研发开辟了令人兴奋的可能性,有望颠覆传统的药物研发模式。
谷歌研究团队希望AlphaFold3能够帮助我们重新认识生物世界、重新思考药物发现。值得一提的是,科研工作者可以免费使用AlphaFold3的大部分功能(https://www.alphafoldserver.com/)。为了进一步挖掘AlphaFold3在药物开发和设计方面的潜力,Isomorphic Labs已经与制药公司合作,将它应用于现实世界的挑战,最终开发出治疗人类最致命疾病的新疗法。
DeepMind对结构生物学的颠覆史
过去10年来,谷歌DeepMind团队在生命科学(特别是结构生物学)领域不断创造新的奇迹。该团队创造这些奇迹的主要利器就是人工智能和深度学习。而这可能要从他们在围棋领域的开创性工作说起。
2016年1月,DeepMind团队在国际著名杂志《Nature》发表了基于人工智能的围棋新算法(AlphaGO)。同年3月,AlphaGO 以4:1的比分,击败韩国职业九段棋手李世石;次年5月,更新版的AlphaGO以3:0的比分,战胜当时世界排名第一的围棋棋手柯洁。这一系列胜利掀开了围棋历史的新篇章,彻底改变围棋训练和竞赛的模式(图2)。

图2. DeepMind开发的AlphaGo第一次战胜人类顶尖围棋棋手李世石九段。
随后,DeepMind转战生物科学领域。
2018年,DeepMind利用卷积神经网络技术,训练出第一代AlphaFold(俗称 AlphaFold1)。AlphaFold1于2018年5月参加第13届国际蛋白质结构预测(CASP)大赛,并且取得不俗战绩。此时的AlphaFold虽然超出了人们的预期,但是与其他顶级研究团队(包括密歇根大学张阳教授团队)相比,并没有明显的优势。在不同的评价指标和范畴下(比如基于模板的TBM蛋白组,图3),其他团队甚至超过AlphaFold1的成绩 (Groups Analysis: zscores - CASP13)。AlphaFold1论文于2020年1月在《Nature》上发表。

图3. DeepMind开发的AlphaFold1第一次参加国际蛋白质结构预测CASP13。此图是CASP13基于模版的TBM蛋白组预测结果,其中密歇根大学张阳团队(Zhang)的整体结构预测精度高于AlphaFold1 (A7D) 的预测精度。
2020年,DeepMind发布第二代AlphaFold(也称AlphaFold2),并参加2020年的第14届CASP大赛。AlphaFold2引进新的Transformer网络,首次实现端到端的机器模型训练。在CASP14上,AlphaFold2在所有的结构模型范畴,甚至是在几乎所有的单个蛋白目标,都遥遥领先于其他参赛团队。这也是DeepMind在生命科学领域,第一次惊艳所有科学家的眼睛,真正的显示了人工智能在解决复杂生命科学上的巨大威力。尽管如此,此时的AlphaFold仅限于蛋白质单链的结构预测,对更加复杂、也更加重要的生物大分子复合物的结构预测尚未涉足。AlphaFold2论文于2021年7月发表在《Nature》期刊上。

图4. DeepMind开发的AlphaFold2第二次参加国际蛋白质结构预测CASP14。AlphaFold2的结果预测精度远远超出所有其他参赛团队的预测结果。这是AI第一次在生命科学领域展现巨大威力。
2021年10月,DeepMind团队将AlphaFold2进一步推广到AlphaFold-Multimer,对蛋白质-蛋白质复合物的四级结构进行预测。尽管突破了该领域的传统,但是AlphaFold-Multimer在复合物的精度只达到了23%(即可以对23%的复合物产生高精度预测结构,改进后版本达到36%),比它在单链蛋白质上所创造的2/3左右的高精度蛋白预测成功率仍然逊色不少。显示出,蛋白质复合物的结构在当时仍然是一个远没有被解决的问题。AlphaFold-Multimer于同期发布在bioRxiv预印本网站上。

图5. 2021-2022年,DeepMind在未经同行审议的预印本网站bioRxiv发布AlphaFold-multimer,预测蛋白质-蛋白质相互作用的复合物结构。
2023 年11月,笔者曾撰文介绍DeepMind研究团队当时发布的最新一代 AlphaFold (论文称之为AlphaFold-last,但是考虑到该模型的进展我们称其为AlphaFold3)。该研究显示其最新的模型现在可以对蛋白质数据库 (PDB) 中的几乎所有分子类型进行预测,通常达到原子精度。
图6. 笔者于2023年11月在CSDN社区第一次介绍AlphaFold3的初步结果(原文链接:https://blog.csdn.net/weixin_4528312/article/details/134238052)。
鉴于AlphaFold的强大能力,谷歌从DeepMind拆分出了一家名为Isomorphic Labs的新药研发公司,致力于使用人工智能加速药物研发。
2024年5月9日(也就是今天),DeepMind与Isomorphic Labs的研究人员在 《Nature》 期刊发表了题为:Accurate structure prediction of biomolecular interactions with AlphaFold 3 的研究论文。该研究正式发布AlphaFold 3程序和研究算法,以前所未有的精确度成功预测了所有生命分子(蛋白质、DNA、RNA、配体等)的结构和相互作用。与现有的预测方法相比,AlphaFold 3 发现蛋白质与其他分子类型的相互作用至少提高了 50%,对于一些重要的相互作用类别,预测准确率甚至提高了一倍。
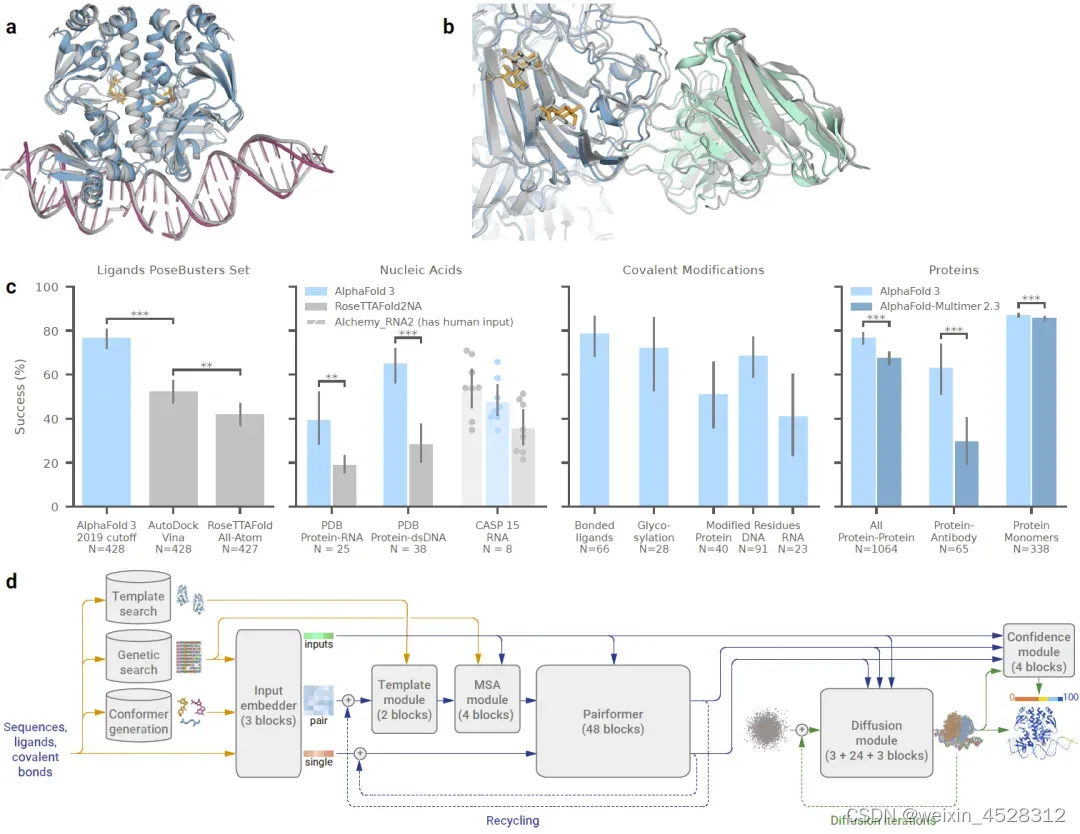
图7. DeepMind 于2024年5月在《Nature》期刊正式发布AlphaFold3的算法和对复合物四级结构的预测结果。
原理:AlphaFold2的模型训练架构
AlphaFold 3 (AF3) 的整体结构与AlphaFold 2 (AF2)相似。他们都有一个大的主干,用于训练出化学复合物的成对表示,然后是一个结构模块,利用这种成对表示生成显式的原子位置。但是每个主要组件都有很大的不同(图8)。AF3的这些新的修改,既是为了满足广泛的化学实体而不过度特殊处理,也是基于对AlphaFold 2在不同修改下性能的观察。

图8. AlphaFold2和AlphaFold3的框架结构比较。(A) AlphaFold2;(B)AlphaFold3。
AF3对AF2的改进主要是在以下四个方面:
(1)减少MSA模块的影响
在AF3的主干模块中,MSA处理被大幅减少,采用了更小更简单的MSA嵌入块。与AF2的原始Evoformer相比,块的数量减少到四个,MSA表示的处理采用了廉价的成对加权平均,后续处理步骤仅使用了成对表示。"Pairformer"取代了AlphaFold 2的"Evoformer"成为主要的处理块(图9)。它仅操作成对表示和单一表示;MSA表示未保留,所有信息都通过成对表示传递。成对处理和块的数量基本与AlphaFold 2相同。

图9. AlphaFold3的Pairformer模块。输入和输出:维度为 (n, n, c) 的配对表示和维度为 (n, c) 的单一表示。 n:token 数量(聚合物残基和原子),c:通道数量(配对表示为 128,单一表示为 384)。 48 个块中的每一个都有一组独立的可训练参数。
(2)引入扩散模块,取代AF2结构模块
AF3的主干模块得到的成对和单一表示与输入表示后,一起传递给新的扩散模块。这种新的扩散模块取代了AlphaFold 2的结构模块。和AF2的结构模块不同,AF3的扩散模块不需要旋转框架或任何等变处理,它直接在原子坐标和粗粒化的表示上进行构象学习。这可能是和AF2相比,AF3最大的创新之处。
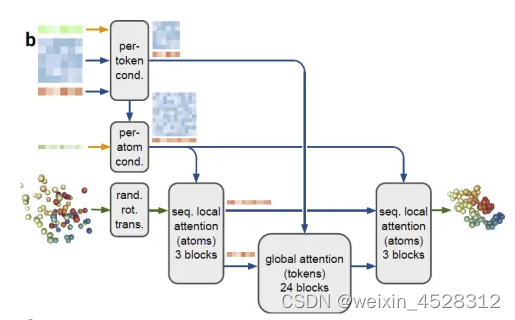
图10. AlphaFold3的扩散(diffusion)模块。输入:粗数组表示每个 token 的表示(绿色:输入,蓝色:对,红色:单个)。细数组表示每个原子的表示。彩色球表示物理原子坐标。
DeepMind的作者在AlphaFold 2中就已经观察到,消除大部分结构模块的复杂性对预测准确性只有较小影响,而保持主链框架和侧链扭转表示对一般的分子图增加了相当多的复杂性。类似地,AlphaFold 2在训练期间需要精心调整的立体化学违规惩罚来强制执行结果结构的化学合理性。因此,作者使用相对标准的扩散方法,在这种方法中,扩散模型被训练接收“加噪”的原子坐标然后预测真实坐标(图10)。
这个任务要求网络在多种长度尺度上学习蛋白质结构,其中小噪声下的去噪任务强调了对局部立体化学的理解,而高噪声下的去噪任务则强调了系统的大尺度结构。在推断时,会采样随机噪声,然后通过递归去噪产生最终的结构。重要的是,这是一个生成式的训练过程,会生成一系列答案的分布。这意味着,对于每个答案,局部结构都将被明确定义,即使网络对位置不确定。因此,作者能够避免对残基进行基于扭转的参数化和结构上的违规损失,同时处理通用配体的全部复杂性。与最近的一些工作类似,作者发现,与分子的全局旋转和平移相关的不变性或等变性在体系结构中是不需要的,因此作者省略了它们,以简化机器学习架构。

图11. AlphaFold3的扩散(diffusion)模块。输入:粗数组表示每个 token 的表示(绿色:输入,蓝色:对,红色:单个)。细数组表示每个原子的表示。彩色球表示物理原子坐标。
(3)交叉蒸馏法
采用生成式扩散方法也带来了一些技术挑战。最大的问题是生成模型容易产生幻觉,即模型可能在无结构的区域中发明看似合理的结构。为了抵消这种效果,作者使用了一种新颖的交叉蒸馏方法,其中作者通过AlphaFold-Multimer v2预测的结构丰富了训练数据。在这些结构中,无结构区域通常由长的延伸环代替紧凑的结构,并在它们上进行训练可以使AlphaFold 3模仿这种行为。这种交叉蒸馏大大减少了AF3的幻觉行为。
(4)置信度回滚
作者进一步开发了置信度度量,用于预测最终结构中的原子级和成对级误差。在AlphaFold 2中,这是通过在训练期间回归结构模块输出中的误差来直接完成的。然而,这个过程对扩散训练不适用,因为扩散训练只训练了一个扩散步骤,而不是一个完整的结构生成。为了解决这个问题,作者开发了一种扩散的“回滚”程序,用于训练期间的完整结构预测生成。然后使用这个预测的结构对对称的真实链和配体进行排列,并计算性能指标来训练置信度头。置信度头使用成对表示来预测LDDT(pLDDT)和预测对齐误差(PAE)矩阵,就像AlphaFold 2中一样,以及距离误差矩阵(PDE),即与真实结构相比预测结构的距离矩阵中的误差。
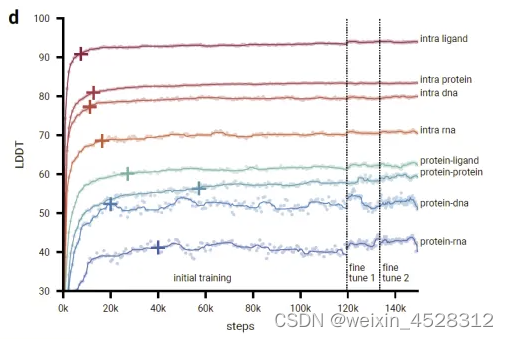
图12. AlphaFold3初始训练和微调阶段的训练曲线,显示了作者评估集上的 LDDT 作为优化器步骤的函数。散点图显示原始数据点,线条显示使用核宽度为 9 个数据点的中值滤波器的平滑性能。十字标记平滑性能达到其初始训练最大值的 97% 的点。
图12显示,在初始训练期间,模型迅速学习预测局部结构(所有链内指标迅速上升,并在前20000个训练步骤内达到最大性能的97%),而模型需要较长时间来学习全局结构(界面指标增长缓慢,并且蛋白质-蛋白质界面LDDT仅在60000个步骤后才达到97%)。在AF3的开发过程中,作者观察到一些模型能力相对较早达到顶峰并开始下降(很可能是由于对这种能力的有限训练样本过拟合),而其他能力仍然未被充分训练。因此,作者通过增加/减少相应训练集的采样概率以及使用所有上述指标的加权平均和一些额外指标进行早停来解决这个问题,以选择最佳模型检查点。使用更大的裁剪大小进行的微调阶段改进了所有指标的模型,特别是在蛋白质-蛋白质界面上有很大提升。
结果:
(1) AF3对不同复合物类型的预测精度
AF3可以从输入的聚合物序列、残基修饰和配体SMILES中预测结构。图13中展示了一些示例,突显了该模型对多种生物学重要和治疗相关模态的泛化能力。选择这些示例时考虑了个体链和界面与训练集的相似性的新颖性。

图13. AlphaFold3 复合物结构预测的的示例。 预测的蛋白质链以蓝色显示(预测的抗体以绿色显示),预测的配体和聚糖以橙色显示,预测的 RNA 以紫色显示,实验结构以灰色显示。(a) 人体40S 小核糖体亚基(7663 个残基,PDB ID 7TQL, GDT 86.9)。(b)EXTL3 同型二聚体的糖基化球状部分(PDB ID 7AU2,RMSD:1.10 Å)。 (c)间皮素 C 末端肽与单克隆抗体 15B6 结合(PDB ID 7U8C,DockQ:0.85)。 (d)临床阶段抑制剂LGK974与PORCN结合,与WNT3A肽形成复合物(PDB ID 7URD,配体RMSD 1.00 Å)。 (e)(5S,6S)-O7-磺基DADH以新折叠方式与AziU3/U2复合物结合(PDB ID 7WUX,配体RMSD 1.92 Å)。 (f) NIH-12848的类似物与PI5P4Kγ的变构位点结合(PDB ID 7QIE,配体RMSD 0.37 Å)。
(a) 蛋白质-配体结构预测(PoseBusters基准集):
作者仔细评估了该系统在每种复合物类型的最新界面特定基准上的性能。对于蛋白质-配体界面的性能评估是在PoseBusters基准集上进行的,该集合由2021年或之后发布到PDB的428个蛋白质-配体结构组成。由于AF3的标准训练截止日期是在2021年,研究人员单独训练了一个带有较早训练集截止日期的AF3模型。在PoseBusters集合上的准确度报告为蛋白质-配体对中口袋对齐配体RMSD小于2 Å的百分比。
基准模型分为两类:一类仅使用蛋白质序列和配体SMILES作为输入,另一类则额外泄露了已解析的蛋白质-配体测试结构的信息。传统的对接方法使用后者的特权信息,尽管在实际使用情况下这些信息不可用。即便如此,AlphaFold 3在不使用任何结构输入的情况下也大大优于诸如Vina等经典对接工具(Fisher精确p=2.27 * 10-13),甚至在不使用任何结构输入的情况下也远远优于所有其他真正的盲对接工具,如RoseTTAFold All-Atom(p=4.45 * 10-25)。文章的扩展数据图3显示了三个示例,其中AlphaFold 3实现了准确预测,但对接工具Vina和Gold没有成功。
PoseBusters分析是使用了2019年9月30日的AlphaFold 3训练截止日期,以确保模型未经过PoseBusters结构的训练。为了与RoseTTAFold All-Atom的结果进行比较,作者使用了PoseBusters版本1。同时,作者使用多个种子以确保正确的立体化学并避免轻微的蛋白质-配体碰撞(而不是像扩散引导一样强制执行),但通常能够产生高质量的立体化学。另外,作者还训练了一个接收“口袋信息”的AlphaFold 3版本,这是一些最近深度学习工作所使用的。
(b) RNA-蛋白质复合物预测:
AF3能够比RoseTTAFold2NA更准确地预测蛋白质-核酸复合物和RNA结构。由于RoseTTAFold2NA仅针对1000个残基以下的结构进行验证,研究人员仅使用最近的PDB评估集中1000个残基以下的结构进行比较。AlphaFold 3能够预测具有数千个残基的蛋白质-核酸结构,图13a显示了其中的一个示例。请注意,研究人员没有直接与RoseTTAFold All-Atom进行比较,但基准表明,RoseTTAFold All-Atom在核酸预测方面与RoseTTAFold2NA相当或略低于其准确性。研究人员还评估了AF3在10个公开可用的CASP15 RNA目标上的表现:研究人员在自己和它们的预测的公共子集上的平均性能高于RoseTTAFold2NA和AIchemy_RNA(CASP15中的最佳基于AI的提交,也是最好的AI)。
在各自的常见子集上,AF3没有达到最佳人类专家辅助的CASP15提交AIchemy_RNA241的性能水平。由于数据集大小有限,研究人员在正文中没有报告显著性检验统计数据。有关仅预测核酸(不包括蛋白质)的准确性的进一步分析,在论文的扩展数据图5b有展示。
(c) 共价修饰预测:
AF3还可以准确预测共价修饰(键合配体、糖基化和修饰的蛋白质残基和核酸碱基)。这些修饰包括任何聚合物残基(蛋白质、RNA或DNA)。作者对键合配体和糖基化数据集应用质量过滤器(与PoseBusters一样):他们仅包括具有高质量实验数据的配体(根据RCSB结构验证报告,ranking_model_fit > 0.5,即具有中位数以上模型质量的X射线结构)。与PoseBusters集合一样,键合配体和糖基化数据集不是通过与训练数据集的同源性来筛选的。基于结合的聚合物链同源性的筛选(使用聚合物模板相似度 < 40)只产生了5个键合配体和7个糖基化的聚类。在这里,作者排除了多残基糖基,因为RCSB验证报告没有为它们提供ranking_model_fit值。在所有质量实验数据上,多残基糖基的成功预测百分比(口袋RMSD < 2 Å)为42.1%(N = 131聚类),略低于所有质量实验数据上单残基糖基的成功率46.1%(N = 167)。修改的残基数据集与其他聚合物测试集类似进行了过滤:它仅包含具有与训练集低同源性的聚合物链中的修改残基。
(d) 蛋白质-蛋白质复合物结构预测:
在扩展建模能力的同时,相对于AlphaFold-Multimer v2.3,AF3在蛋白质复合物准确性上也有所提高。通常,蛋白质-蛋白质预测成功率(DockQ > 0.23)增加了(配对Wilcoxon符号秩检验,p = 1.8 * 10-18),特别是抗体-蛋白质相互作用的预测表现出显著改善。单体蛋白质LDDT的改善也是显著的。AF3对MSA深度的依赖性与AF-M 2.3非常相似;具有浅MSA的蛋白质预测准确性较低。
(2) AF3模型的置信度与真实准确性的关联
与AlphaFold 2类似,AlphaFold 3的置信度度量与准确性很好地校准。作者的置信度分析是在最近的PDB评估集上进行的,没有同源性过滤,包括肽段。他们对配体类别进行了过滤,只考虑高质量的实验结构,并且只考虑标准的非键合配体。所有统计数据都是按聚类加权的,并仅考虑排名最高的预测。
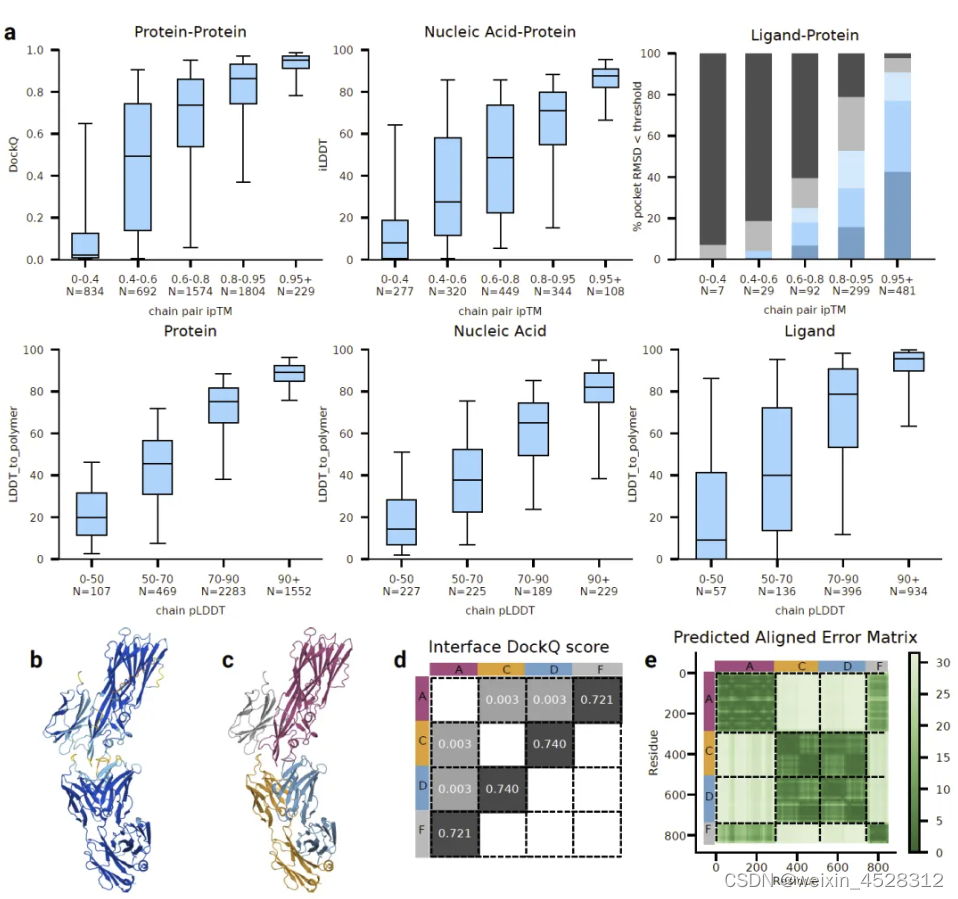
图14. AlphaFold 3 模型预测的置信度。 (a)(上部),含蛋白质界面的准确度作为链对 ipTM 的函数。 a(下部),LDDT_to_polymer 的准确度针对各种链类型进行评估,作为链平均 pLDDT 的函数。箱线、中心线和晶须边界分别位于(25%、75%)间隔、中位数和(5%、95%)间隔。 N 值报告每个带中的簇数。 (b)PDB ID 7T82 的预测结构由 pLDDT 着色(橙色:0-50,黄色:50-70,青色 70-90 和蓝色 90-100)。 (c)相同的预测按链着色。 (d)蛋白质-蛋白质界面的 DockQ 分数。 (e)相同预测的预测对齐误差 (PAE) 矩阵(颜色越深表示越可信),侧边栏上为面板 c 的链着色。黑色虚线表示链边界。
图14a的上半部,作者将链对ipTM(界面预测TM-score)与界面准确性度量绘制出来:蛋白质-蛋白质DockQ,蛋白质-核酸iLDDT以及蛋白质-配体成功,成功定义为在经过阈值处理的口袋对齐RMSD值下的例子百分比。在图14a的下半部,作者将每个蛋白质、核苷酸或配体实体的平均pLDDT绘制出来,与专门设计的LDDT_to_polymer指标进行比较,该指标与pLDDT预测器的训练目标密切相关。
图14b-e中,研究人员突出显示了7T82的单个示例预测,其中每个原子的pLDDT着色标识了不确定的链尾、有些自信的界面以及其余自信的次级结构。在图14c中,相同的预测按链进行了着色,图14d显示了DockQ界面评分,图14d的轴上显示了每个链的颜色作为参考。从图14e中我们可以看到,对于DockQ > 0.7的粉灰色和蓝橙色残基对,PAE置信度很高,而对于具有DockQ≈0的粉橙色和粉蓝色残基对,置信度最低。在另一个示例中,含有蛋白质和核酸链的PAE分析请参见论文扩展数据图5c-d。
(3) AlphaFold3 模型局限性
就像任何尖端技术一样,AlphaFold3也有它自己的局限性。AlphaFold 3模型主要在立体化学、幻觉、动态性和对某些目标的准确性方面存在局限性。
(a)AF3模型可能存在手性错误
立体化学方面,研究人员注意到两个主要的违规类别。首先,尽管模型接收了具有正确手性的参考结构作为输入特征,但AF3模型的输出并不总是遵守手性规则。为了解决这个问题,在PoseBusters基准测试中,研究人员在模型预测的排名公式中包含了手性违规的惩罚项。尽管如此,研究人员仍然观察到基准测试中4.4%的手性违规率。第二类立体化学违规是模型偶尔会产生重叠(“碰撞”)的原子预测。有时这会表现为同构体中的极端违规,其中整个链被观察到重叠。在排名时对碰撞进行惩罚可以减少这种故障模式的发生,但无法完全消除。几乎所有剩余的碰撞都发生在蛋白质-核酸复合物中,这些复合物既有超过100个核苷酸又有超过2000个残基。
其次,研究人员注意到从非生成式的AlphaFold 2模型转换为基于扩散的AlphaFold 3模型引入了在无序区域产生虚假结构顺序(幻觉)的挑战。虽然幻觉区域通常被标记为非常低的置信度,但它们可能缺乏AlphaFold 2在无序区域产生的独特的丝带状外观。为了在AF3中鼓励产生丝带状预测,作者使用了AlphaFold 2预测的蒸馏训练,并添加了一个排名项以鼓励产生更多的溶剂可及表面积。

图15. AF3模型的一些局限性。(a)抗体预测质量随模型种子数量的增加而增加。排名靠前、同源性较低的抗体-抗原界面预测的质量是种子数量的函数。每个数据点显示 1200 个种子中 1,000 个种子随机样本(有替换)的平均值。置信区间是每个数据点 10,000 次聚类得分重采样的 95% 引导。每个界面的样本按蛋白质-蛋白质 ipTM 排序。显着性检验采用双侧 Wilcoxon 符号秩检验。N = 65 个簇。*** 表示 p< 0.001。P 值:2.0 * 10-5 表示正确率,p=0.009 表示非常高准确率。 (b)PoseBusters 集 (PDB ID 7CTM) 中的目标 Thermotoga maritima α-葡萄糖醛酸酶和 β-D-葡萄糖醛酸的预测 (彩色) 和基本事实 (灰色) 结构。 AF3 预测 α-D-葡萄糖醛酸,不同的手性中心用星号表示。所示的预测按配体蛋白 ipTM 排名靠前,并具有手性和冲突惩罚。 (c)构象覆盖范围有限。 cereblon 的基本事实结构 (灰色) 处于开放 (apo PDB ID 8CVP,左) 和封闭 (holo mezigdomide 结合,PDB ID 8D7U,右) 构象。 apo (有 10 个重叠样本) 和全息结构的预测 (蓝色) 均处于封闭构象。虚线表示 N 端 Lon 蛋白酶样和 C 端沙利度胺结合域之间的距离。 (d)具有 1,854 个未解析残基的核孔复合物(PDB ID 7F60)。真实值(左)和来自 AF-M 2.3(中)和 AF3(右)的预测。(e)具有重叠 DNA(粉色)和蛋白质(蓝色)链的三核小体的预测(PDB ID 7PEU);突出显示的是重叠的蛋白质链 B 和 J 以及自重叠的 DNA 链 AA。
(b)AF3不能预测动态行为
蛋白质结构预测模型的一个关键局限性是它们通常预测的是PDB中所见的静态结构,而不是溶液中生物分子系统的动态行为。这个限制在AlphaFold 3中仍然存在,即使使用多个随机种子来进行扩散头或整个网络的训练,也无法产生解集的近似。
某些情况下,模拟的构象状态可能不正确或不全面,鉴于指定的配体和其他输入。例如,E3泛素连接酶在天然状态下采用开放构象,只有当结合配体时才观察到闭合状态,但是AF3专门预测了闭合状态,无论是在完整还是天然系统中。许多方法已经被开发出来,特别是围绕MSA重新采样,它们有助于从先前的AlphaFold模型中生成多样性,并且也可能有助于使用AF3进行多状态预测。
(c)模型甄选和排名有待提高
为了获得最高的准确性,可能需要生成大量的预测并对其进行排名,这会增加额外的计算成本。研究人员观察到这种效应最明显的一类目标是抗体-抗原复合物,与其他近期的工作类似。图15a显示,对于AlphaFold 3,随着模型种子数量的增加,排名靠前的预测结果不断提高,甚至在1000个种子时仍然如此(使用蛋白质-蛋白质界面ipTM进行排名)。在其他类别的分子中,一般不会观察到使用多个种子时的这种大幅提高。对于AF3预测,每个模型种子只使用一个扩散样本而不是五个并不会显著改变结果,这表明运行更多的模型种子对于抗体评分的改进是必要的,而不仅仅是增加扩散样本。
因此,尽管AlphaFold 3在建模精度方面取得了巨大进步,但仍有许多目标的精确建模可能具有挑战性。
总结:
分子生物学的核心挑战是理解并最终调控生物系统中复杂的原子相互作用。AlphaFold 3模型朝着这个方向迈出了重要的一步,展示了在一个统一的框架中准确预测各种生物分子系统的结构是可能的。
- 虽然在实现所有类型的相互作用的高度准确预测方面仍存在着重大挑战,但AF3的成功证明了可以构建一个深度学习系统,它对所有这些相互作用都表现出强大的覆盖和泛化能力。
- 研究人员还证明了缺乏跨实体进化信息并不是阻碍在预测这些相互作用方面取得进展的重大障碍,而且抗体结果的显著改进表明AlphaFold衍生的方法能够在不依赖MSA的情况下对分子相互作用类别的化学和物理进行建模。
- 最后,蛋白质-配体结构预测的大幅改进表明,在通用深度学习框架内,处理化学空间的广泛多样性是可能的,而无需人为地将蛋白质结构预测与配体对接分开。
- 自下而上地建模细胞组分是解开细胞内分子调控复杂性的关键一步,AlphaFold 3的性能表明,开发正确的深度学习框架可以大大减少获得这些任务上生物相关性能所需的数据量,并放大已经收集到的数据的影响。
研究人员预计,结构建模将继续改进,不仅是因为深度学习的进步,还因为实验结构确定方法的持续方法学进步,比如冷冻电子显微镜和断层扫描的显著改进,将为这些AI模型的泛化能力提供丰富的新训练数据。随着深度学习技术的不断发展,以及实验结构测定方法论的不断革新,基于AI的结构建模将继续进步,将会推动我们进入一个结构信息驱动的生物理解和治疗开发的时代。

图16. AlphaFold3的主要领导者、来自谷歌DeepMind团队的Demis Hassabis博士(左)和 John Jumper博士(右)
参考文献:Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3 | Nature